No suelen generar grandes despliegues mediáticos, ni son el tema central de las charlas de café. Tomadas por separado, las enfermedades poco frecuentes (EPoF) hacen honor a su nombre y son también ‘poco conocidas’. Sin embargo, en conjunto son más de 8.000, y se estima que en Argentina afectan a 1 de cada 13 personas e impactan directamente en promedio sobre una de cada 4 familias.
Su presentación es diversa: algunas comienzan en los
primeros meses de la vida, otras en la adultez; unas cuentan con una
manifestación física visible y otras no. Sin embargo, la mayoría tiene algunas
características comunes: la sospecha de que ‘algo no anda bien’, la consulta a
numerosos médicos que no logran dar con el diagnóstico y los años transcurridos
hasta conocer qué enfermedad se padece, años perdidos durante los cuales se
podría haber iniciado algún tratamiento para mejorar el pronóstico y la calidad
de vida.
Para ser denominada como EPoF, una patología debe
presentarse en menos de 1 cada 2.000 personas. Hoy, 28 de febrero -si fuera año
bisiesto, sería el 29, el día menos frecuente- se conmemora el Día Mundial de
las Enfermedades Poco Frecuentes. “Desde la Fundación Enhué, trabajamos
permanentemente para acompañar a aquellos que padecen una EPoF y a sus
familias, con el objetivo de que su calidad de vida sea la mejor posible. A su
vez, buscamos generar conciencia para que los pacientes puedan llegar al
diagnóstico lo antes posible”, destacó Belén González Sutil, Directora
Ejecutiva de la Fundación Enhué.
---------------------------------------------------------------------------------------------------------------
Alejandro Di Pietro -
AEH
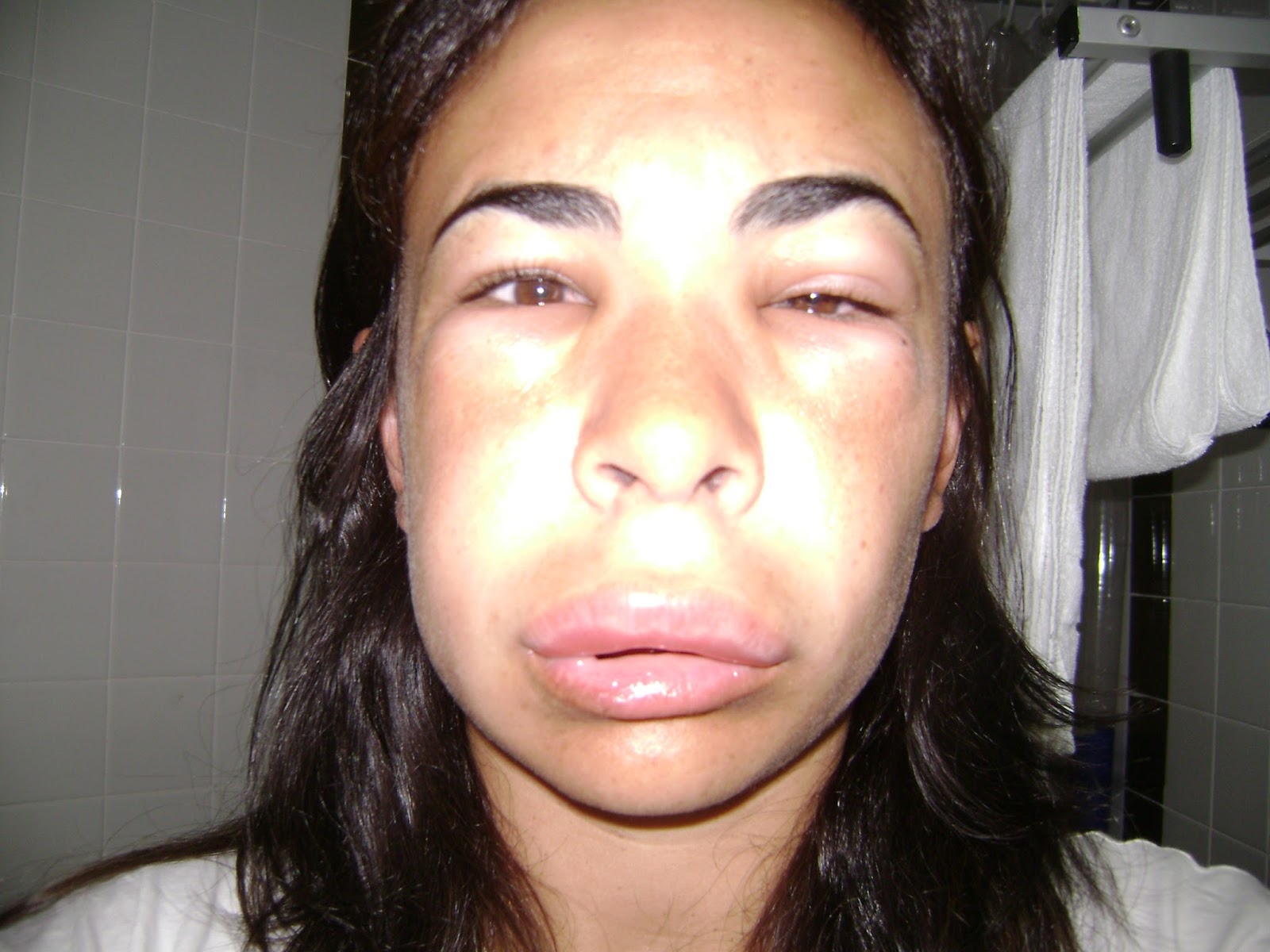
“Mi esposa sufría con mucha frecuencia episodios de
hinchazón y dolores estomacales. Lo tomábamos como algo alérgico y visitamos 3
alergistas distintos, hasta que terminamos en un importante centro
especializado en alergias. Siempre el diagnóstico fue alergia”. Es el relato de
Alejandro, papá de Laureana, una paciente con AEH.
“Luego nació nuestra hija, Laureana, y cuando ella tenía
apenas 1 mes vida mi esposa tuvo un episodio de laringe y la internamos. A las
pocas horas regresamos a casa, pero empeoró y volvimos a internarla y
lamentablemente falleció a los 30 años de edad debido a no estar correctamente
diagnosticada y no contar con la medicación para el angioedema hereditario. La
autopsia refirió un paro cardiorrespiratorio”. Su hermano dos años antes
también había fallecido a los 26 años en un hospital del interior sin que
supieran la causa.
“Cuando le conté lo ocurrido a nuestro alergista, sospechó
por primera vez que podíamos estar ante la presencia de un caso de angioedema
hereditario, y nos derivó con un especialista para estudiar a Laureana, cuyo
examen de laboratorio dio positivo”.
“Hoy Laureana con sus 10 años convive con la enfermedad,
pero tenemos diagnóstico, estamos informados, siempre llevamos nuestra
medicación (seguro de vida) a todos lados, aprendí a aplicársela por si algún
centro asistencial se niega a hacerlo ya que muchos no conocen la enfermedad; con
el tiempo Laureana fue reconociendo los síntomas y hoy manejamos la situación
con tranquilidad y confianza. Hasta conseguimos que la obra social genere un
protocolo de aplicación de la medicación frente a la emergencia”.
El angioedema hereditario (AEH) es una enfermedad genética
que se caracteriza por la aparición de episodios recurrentes de hinchazón o
edemas en distintas partes del cuerpo con compromiso de piel y mucosas, que
además de alterar significativamente la calidad de vida de los pacientes puede
ser potencialmente mortal.
El Dr. Ricardo Zwiener, médico especialista en Alergia e
Inmunología Clínica, describió que “el síntoma principal es el edema que afecta
la cara, extremidades, tracto gastrointestinal, genitales y vía aérea superior
(lengua, faringe y laringe). Los
episodios son recurrentes, se desarrollan de forma gradual y con una duración
de 2 a 5 días. A su vez, son impredecibles y variables en frecuencia,
localización y severidad. En la mayoría de los casos, se presenta en edades
tempranas de la vida (infancia y adolescencia). Afecta a todas las razas y a
ambos sexos”.
“Si un episodio ocurre en la laringe, la hinchazón puede
generar que se ocluya el pasaje de aire y que el paciente muera por asfixia;
por esta razón, ésta es la manifestación más aguda y se trata de una emergencia
médica”, alertó Alejandra Menéndez, paciente con AEH y Presidenta de la
Asociación Argentina de Angioedema Hereditario.
Por otra parte, cuando los edemas ocurren en las manos y
pies, las personas experimentan incomodidad e imposibilidad de realizar algunas
actividades diarias como conducir, escribir, bañarse o peinarse. Los
gastrointestinales producen dolores abdominales fuertes, acompañados de
náuseas, vómitos y diarrea. “Puede confundirse con un cuadro de abdomen
quirúrgico semejante al de una apendicitis o colecistitis, pudiendo llevar a
cirugías abdominales exploratorias innecesarias hasta en un 30% de los
pacientes”, subrayó el Dr. Zwiener, médico de planta del Servicio de Alergia e
Inmunología del Hospital Austral.
El promedio de tiempo de diagnóstico en nuestro país es de
aproximadamente 13 años desde el inicio de los síntomas. Esto se debe a que,
como ocurre con la mayoría de las EPoF, el AEH suele pasar desapercibido para
muchos profesionales de la salud. A su vez, es confundido en numerosas
ocasiones con cuadros alérgicos, por lo que los pacientes pasan muchos años
consultando a distintos especialistas como clínicos, pediatras, alergistas,
emergentólogos, gastroenterólogos y otorrinolaringólogos, hasta llegar a su
diagnóstico final.
Existen terapias para el tratamiento agudo, de emergencia,
orientadas a tratar inmediatamente al paciente cuando sufre un episodio de
edema. Los episodios agudos no responden a la medicación antialérgica como
corticoides, antihistamínicos ni adrenalina: se resuelven únicamente con la
medicación específica. Por este motivo, los especialistas aseguran que el
diagnóstico temprano, que surge de una sospecha clínica y se confirma con
exámenes de laboratorio o genéticos, es de suma importancia para las personas, quienes
mientras no sepan que padecen esta patología, se encuentran desprotegidas, con
alteración de su calidad de vida y con riesgo de vida. Asimismo, al conocer el
diagnóstico, ya saben qué es lo que les sucede y cómo proceder.
“En los últimos años se han logrado grandes avances de la
enfermedad y somos optimistas de que en un futuro no muy lejano vamos a poder
ofrecer nuevas alternativas terapéuticas y con ello una mejora a los pacientes.
El desarrollo de nuevos agentes ha permitido a los pacientes poder tratarse
ellos mismos en su domicilio sin necesidad de recurrir a una guardia”, concluyó
el Dr. Zwiener.
---------------------------------------------------------------------------------------------------------------
Lorena García - MPS

“Desde el primer momento, yo sabía que Iván tenía algo. En
los controles, sus valores clínicos estaban bien y los médicos me decían que
estaba todo bien, pero yo le explicaba al doctor que algo le pasaba, y más de
uno me dijo que estaba loca, que lo dejara crecer tranquilo. Yo le veía los
brazos muy largos y una cara particular”, describió Lorena, la mamá de Iván,
que ahora tiene 10 años.
“Al año y siete meses, la doctora abrió los ojos y vio que
algo pasaba. Fuimos a un neumonólogo, porque Iván tenía el torso muy ancho.
Luego, lo derivaron a un traumatólogo por su giba, que ya se le notaba.
Finalmente, a los 3 años, le diagnosticaron MPS y luego, en Buenos Aires, nos
especificaron que era tipo II. Fue una gran alegría recibir el diagnóstico;
finalmente supimos qué le pasaba y encima conocimos que tenía tratamiento”.
Las mucopolisacaridosis (MPS) son un grupo de patologías
poco frecuentes de origen genético. Se producen por la acumulación en los
lisosomas de moléculas llamadas mucopolisacáridos, por deficiencia de alguna de
las enzimas que están implicadas en su degradación. Esto genera daños en
diversos tejidos, especialmente en el tejido conectivo hallado en hueso,
cartílago, piel, etc.
Durante el primero y segundo año, entre los signos iniciales
que podrían estar indicando la presencia de un tipo de MPS (la tipo II o
enfermedad de Hunter) se encuentran las otitis recurrentes, secreciones nasales
persistentes y hernias (umbilicales o inguinales). De manera lentamente
progresiva suelen presentarse, entre otros signos, deformidades en la columna y
las manos, articulaciones rígidas, cuello corto, talla baja, cabello grueso,
abdomen grande (por el aumento de tamaño del hígado), dificultades en la vista
y en la audición, y afectación en el desarrollo intelectual.
“La importancia del diagnóstico temprano radica en que por
tratarse de enfermedades progresivas, cualquier tratamiento instalado
precozmente será más efectivo. En algunas MPS se ha desarrollado un tratamiento
de reemplazo de la enzima deficiente,
que atenúa los síntomas de la enfermedad. De esta manera, cuanto antes se
diagnostique, mejor será el pronóstico”, manifestó la Dra. Norma Specola,
médica especialista en Neurología Infantil.
Lamentablemente, se encuentra subdiagnosticada. Al formar
parte de las enfermedades poco frecuentes, casi no son difundidas en los
estudios de pregrado y hay muy pocos especialistas que puedan reconocer los
síntomas, que son de aparición lenta, muy sutiles en el comienzo. Una vez que
son más reconocibles, ya hay un mayor grado de compromiso multisistémico.
La Dra. Specola, también Jefa de la Unidad de Metabolismo
del Hospital de Niños de La Plata, añadió que “el defecto genético genera la
producción deficiente de una enzima. Además del tratamiento sintomático que
debe ser implementado en todos los pacientes con MPS, en la enfermedad de
Hunter puede reponerse la enzima faltante por medio de una infusión endovenosa
cada semana”.
El pronóstico de los pacientes con MPS II bajo tratamiento
depende de cuán precoz sea el diagnóstico. Si el niño es pequeño y los síntomas
son leves, la terapia de reemplazo mejora la motricidad articular y la función
cardíaca y pulmonar. Cuánto más avanzada sea la enfermedad, menor será la
respuesta.
“Cuando era chiquito, no hablaba, no pronunciaba bien. Nos
enteramos de que no escuchaba a los 3 años. Le implantaron diábolos, ahora
escucha muy bien y tiene audífonos. Hoy día lleva una vida normal: va a la
escuela, juega, se trepa a los árboles”.
“Luego de comenzar el tratamiento, se mantuvo estable: sus
órganos están bien. Vivir día a día con Iván, con su enfermedad y tratamientos
ya es algo normal. Si no tuviéramos esta rutina, no sé qué haríamos. No podría
ver a Iván en otra persona: es parte nuestra y no lo cambio por nada. Es un
nene muy feliz, le encanta jugar a la play y bailar”, concluyó Lorena.
---------------------------------------------------------------------------------------------------------------
Alejandro Castro –
Enfermedad de Fabry

“A mí me encanta jugar a la pelota. Cuando era chico y
jugábamos, veía a los demás transpirar como locos y yo, seco, aunque hubiera 40
grados de calor. También sentía dolores que no tenían razón de ser, como una
sensación de quemazón en las manos y los pies.”
“Pasé por un montón de médicos que decían que debía ser
dolor por el crecimiento. Finalmente, a los 13 años, me diagnosticaron gracias
a mi primo. Él se hizo ver por un neurólogo que le confirmó que tenía Fabry y
le dijo que, como es hereditaria, su familia se debía testear también. Si uno
mira mi árbol genealógico, los varones tenían muchísimos problemas de salud:
vivían hasta los 35 años”, contó Alejandro, que hoy tiene 34, trabaja y está en
pareja.
“La enfermedad de Fabry es una patología caracterizada por
la deficiencia de una enzima, que lleva a la acumulación de una sustancia en
los lisosomas de las células. A largo plazo, esto puede perjudicar a diversos
órganos como el corazón, riñones y cerebro, aumentando el riesgo de ACV,
insuficiencia cardíaca o renal crónica e infarto agudo de miocardio. Si no se
realiza el tratamiento correspondiente, se reduce la expectativa de vida en 20
años en los varones y 10 en las mujeres” explicó el Dr. Pablo Neumann, médico
nefrólogo y Jefe del Servicio de Nefrología de IPENSA, La Plata.
Algunos de los síntomas que presenta, como dolor quemante en
pies y manos, pueden llevar a que se la confunda con otras patologías como
fibromialgia, dermato o polimiositis, artritis reumatoidea o fiebre reumática.
Otras manifestaciones pueden ser dolores abdominales, diarreas frecuentes,
falta de transpiración con intolerancia a altas temperaturas, disminución de la
audición (hipoacusia) y mareos. En forma distintiva pueden aparecer depósitos
en la córnea (conocida como córnea verticillata) y manchas de color rojo oscuro
en distintas partes del cuerpo (angioqueratomas), que junto con la proteinuria
y la hipertrofia del ventrículo izquierdo, permiten en muchos casos sospechar
la enfermedad.
“Los pacientes con enfermedad de Fabry deambulan, en
promedio, durante 10 a 12 años por varios consultorios, antes de llegar al
diagnóstico de la enfermedad, lo que genera un importante retraso en el inicio
del tratamiento. Esta variable influye sobre el pronóstico y la calidad de vida
de los pacientes”, agregó el Dr. Neumann, que además es docente en la Facultad
de Medicina de la Universidad Nacional de La Plata.
Actualmente, existe un tratamiento para las personas con
Fabry que consta del reemplazo de la enzima deficiente. Para que su eficacia sea
óptima, se debe arribar al diagnóstico de manera temprana y el tratamiento debe
comenzar lo antes posible.
“Cinco años después del diagnóstico, finalmente llegó el
tratamiento al país y comencé a recibirlo. Al año, ya noté una mejoría: empecé
a transpirar, el dolor mermó y los valores de los estudios mejoraron, lo que es
muy importante. Consta de 1 hora cada 15 días y la adherencia es fundamental”.
Apenas nacieron sus dos hijos, Alejandro les hizo el estudio
para corroborar si padecían la patología. Lamentablemente, su hija de 4 años
dio positivo, pero él no se desanima: “La nena ya va al mismo médico que yo y
él decidirá cuándo debe comenzar a tratarse, pero es una enfermedad como
cualquier otra”. Lo negativo, aclaró, “es que las obras sociales hacen todo lo
posible para no cubrir el tratamiento. Recientemente, comencé un nuevo trabajo
y no me aceptan en las distintas entidades. El tiempo es valioso y el
tratamiento salva vidas, por lo que esa actitud no me parece buena”.
---------------------------------------------------------------------------------------------------------------
Soledad Rodríguez -
Enfermedad de Gaucher


“Mi hijo, Rodrigo, desde que nació tenía anemia. A los 5
años, comenzó a tener moretones feos de la noche a la mañana. Yo le preguntaba
si se había caído de la cama y me decía que no. Era muy flaquito y tenía la
panza hinchada. Mi suegra me dijo que lo hiciera ver, que esos síntomas eran
similares a los de un hijo suyo que había muerto y le habían dicho que era
leucemia. Luego de 4 meses de tratamiento por anemia, lo derivaron a un
hematólogo y, luego de 4 meses más, finalmente tuvimos el diagnóstico”, remarcó
Soledad, mamá de Rodrigo.
“Rodri estaba con muchas ojeras, flaco y con la panza que le
salía. Se diagnosticó a tiempo, pero al límite. Ver cómo tu hijo se deteriora
día a día y no poder hacer nada es muy feo. Por eso, el diagnóstico fue muy
esperanzador. Por fin saber qué era, y encima saber que había un tratamiento
disponible, fue una gran noticia. Antes, se lo veía en un retroceso. A partir
de allí, en un avance”.
Le enfermedad de Gaucher es una patología congénita que
aqueja a 1 en 50.000 personas. Sin embargo, en la población de judíos
askenazíes es mucho más frecuente, afectando a 1 de cada 850 individuos.
Se caracteriza por la
acumulación de sustancias tóxicas dentro de algunas células, causada por
la disminución de la actividad de una enzima. Esto genera aumento del tamaño
del hígado y bazo, disminución de glóbulos rojos y plaquetas, y daño óseo.
Los síntomas son en general de instalación lenta y
progresiva. Cuando se diagnostica la enfermedad, muchos de éstos ya están
presentes desde hace años. Entre los síntomas, se encuentran: anemia, palidez,
sangrados, abdomen “hinchado” y dolores óseos progresivos.
“La sospecha diagnóstica es fundamental siempre. Una buena
indagación sobre la historia clínica y familiar, un correcto examen médico y
estudios de laboratorio e imágenes son fundamentales para orientar el
diagnóstico. En un trabajo publicado en 2007, el Dr. Mistry les presentó un
caso típico de Enfermedad de Gaucher a médicos con experiencia en esta
patología y sólo el 25% pensó en ella como un diagnóstico posible”, señaló el
Dr. Diego Fernández Sasso, médico especialista en pediatría y hematología.
“Mi otra hija, More, presentaba algunos síntomas aislados.
Un día tuvo una caída de 20 centímetros y se quebró el peroné y el cartílago de
crecimiento de la rodilla. Los médicos no nos creían, pensaban que era un tema
de violencia doméstica. Finalmente cuando Rodri se diagnosticó, nos aconsejaron
que se lo hagamos a todos y ahí le dio positivo”.
“A partir del diagnóstico, Rodri dio un vuelco de 180
grados. En el ánimo y en su color, la mejoría fue casi inmediata. En los
valores de los estudios tardó un poco más. El doctor nos recomendó que hicieran
algún deporte. Rodri hace básquet y More nos pidió practicar patinaje
artístico. Nos dio miedo, pero la apoyamos y hoy compite profesionalmente! Los
médicos no lo pueden creer”, remarcó Soledad.
Si bien la mayoría de los síntomas se pueden revertir con
los tratamientos disponibles, el daño óseo es la excepción. De esta manera, el
diagnóstico precoz se torna fundamental, ya que el deterioro de los huesos
provoca limitaciones físicas, psicológicas y sociales que pueden alterar la
calidad de vida de quienes las sufren. Un diagnóstico tardío duplica el riesgo
de padecer estas secuelas invalidantes.
La enfermedad de Gaucher, si no es tratada, afecta
notablemente la calidad de vida: un paciente con anemia está cansado, con dolor
crónico, no puede trabajar ni estudiar bien y con el abdomen hinchado no se
siente cómodo. En los casos más graves, requieren transfusiones y las lesiones
óseas pueden ser completamente invalidantes.
En cuanto al tratamiento, el Dr. Fernández Sasso, Jefe de
Internación Pediátrica del Instituto Argentino de Diagnóstico y Tratamiento
(IADT), aclaró que “hoy existen en nuestro país dos tratamientos de reemplazo
enzimático. Son enzimas recombinantes, una de origen animal y otra de origen
humano. Estas enzimas reemplazan a la enzima insuficiente, logrando que el
paciente mejore los síntomas, permitiéndole en casi todos los casos recuperar
una vida normal”.



